asv_probabilities = c(0.3, 0.2, 0.1, rep(0.05, 3), rep(0.025, 4), rep(0.001, 5), rep(0.0001, 5))
names(asv_probabilities) <- paste("ASV", 1:length(asv_probabilities), sep = "")Mock Community Generation
By Hand
mock
diversity
Creation of test communities
Background
In order to make the differences between the metrics presented on this site more apparent, it would be helpful to have a standard mock community that can be used.
Here, I will make two sets of 3 communities - one set that differs in sequencing depth, and another that differs in evenness.
True community with known abundances
First, I will define a true population that mimics a typical microbial community population that has a “long tail” distribution. Our community will have 20 total members, 10 of which are above 1% abundance, and the other 10 are below 1% in abundance.
Each species is named with “ASV” plus a number that increases with decreasing abundance. Don’t worry if you are not familiar with ASVs are what they mean. For now, just think of them as simply the microbial species names.
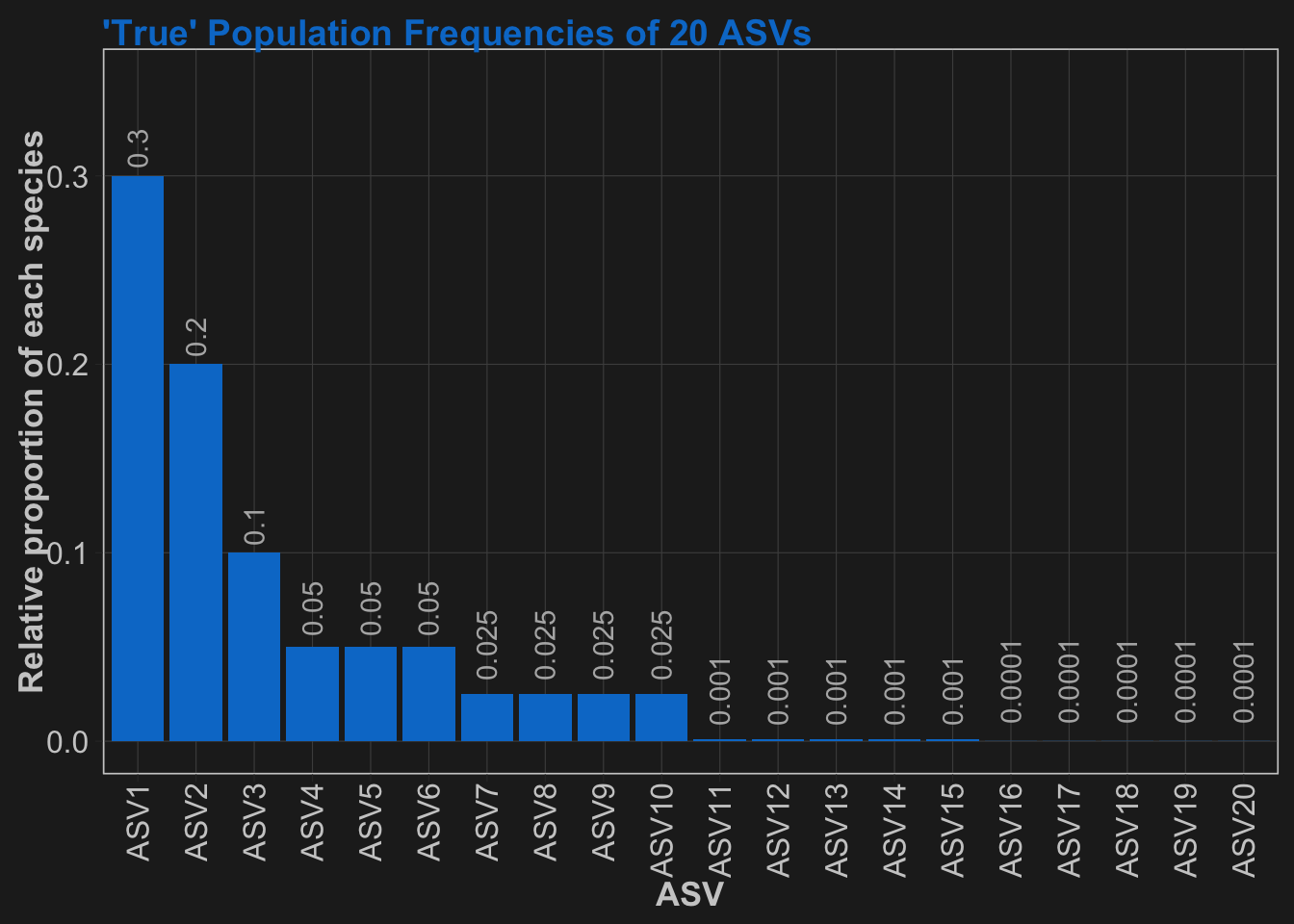
Different sequencing depths (mock community #1)
Starting with this distribution of the true community, let’s take 3 samples at depths of 10,000, 1,000 and 100. This is done with replace=TRUE to simulate a very (infinitely?) large population.
Sample_1 = table(sample(x=names(asv_probabilities), size=10000, replace=TRUE, prob=asv_probabilities))
Sample_2 = table(sample(x=names(asv_probabilities), size=1000, replace=TRUE, prob=asv_probabilities))
Sample_3 = table(sample(x=names(asv_probabilities), size=100, replace=TRUE, prob=asv_probabilities))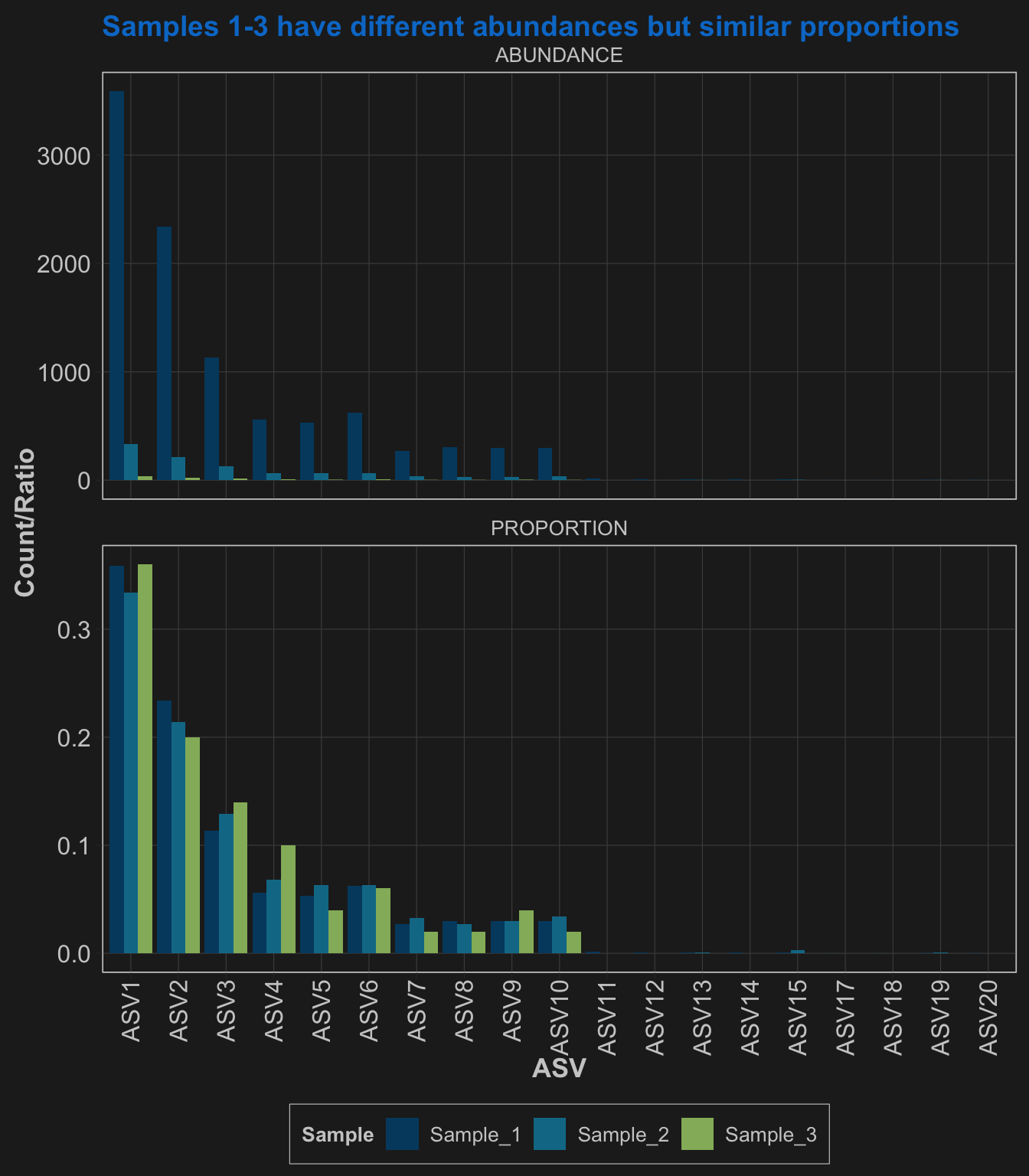
Different diversity/evenness (mock community #2)
Next, we will take these same 20 species and define different true proportions to sample from. These next three communities will have decreasing evenness from 1) all 20 perfectly even, 2) ten species with 10x more abundance than the other 10, and 3) two species with 100x more abundance than the other 18 species.
Then, each will be sampled 10,000 times to give us the same sequencing effort.
Sample_4 = table(sample(x=paste("ASV", 1:20, sep = ""), size=10000, replace=TRUE, prob=c(rep(1,1), rep(1,19))))
Sample_5 = table(sample(x=paste("ASV", 1:20, sep = ""), size=10000, replace=TRUE, prob=c(rep(10,10), rep(1,10))))
Sample_6 = table(sample(x=paste("ASV", 1:20, sep = ""), size=10000, replace=TRUE, prob=c(rep(100,2), rep(1,18))))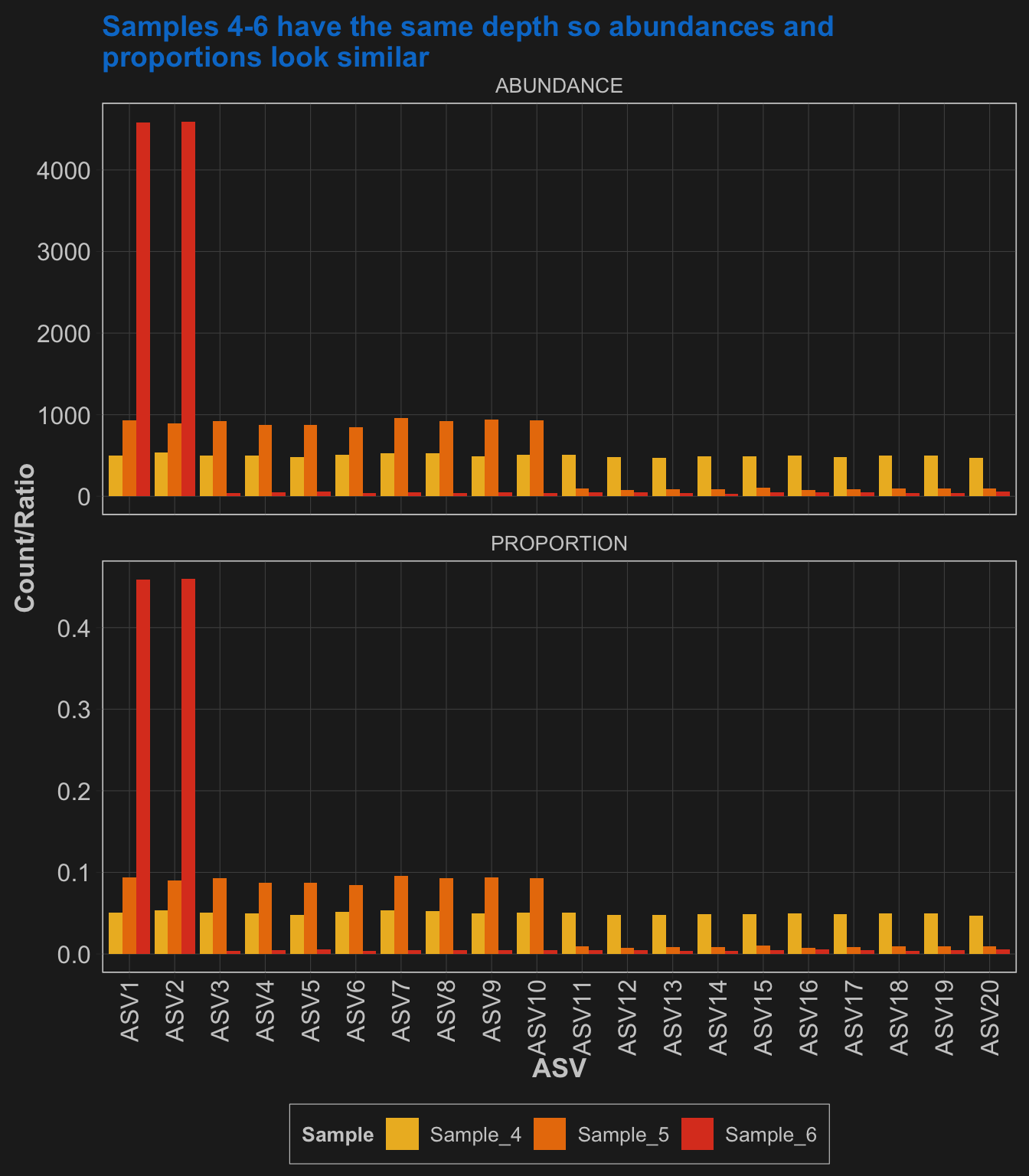
Combining into one dataset
Now, these 6 samples can be made into an “ASV table” with samples in columns, and species names in rows.
d = list(Sample_1, Sample_2, Sample_3, Sample_4, Sample_5, Sample_6)
names(d) = c("Sample_1", "Sample_2", "Sample_3", "Sample_4", "Sample_5", "Sample_6")
mock_community = bind_rows(d, .id = "SAMPLE") %>%
pivot_longer(cols = c(everything(), -SAMPLE), names_to = "ASV") %>%
mutate(value = as.numeric(value), value = ifelse(is.na(value), 0, value)) %>%
pivot_wider(names_from = SAMPLE, values_from = value) %>%
mutate(ASV = fct_relevel(ASV, gtools::mixedsort(unique(as.character(.$ASV))))) %>%
arrange(ASV)| Same Evenness, Differing Depth or Same Depth, Differing Evenness | ||||||
| ASV | Different Depth | Different Evenness | ||||
|---|---|---|---|---|---|---|
| Sample_1 | Sample_2 | Sample_3 | Sample_4 | Sample_5 | Sample_6 | |
| ASV1 | 3589 | 334 | 36 | 503 | 934 | 4584 |
| ASV2 | 2337 | 214 | 20 | 536 | 897 | 4594 |
| ASV3 | 1134 | 129 | 14 | 504 | 926 | 42 |
| ASV4 | 559 | 68 | 10 | 498 | 875 | 47 |
| ASV5 | 533 | 63 | 4 | 480 | 873 | 56 |
| ASV6 | 624 | 63 | 6 | 513 | 848 | 40 |
| ASV7 | 268 | 33 | 2 | 531 | 955 | 50 |
| ASV8 | 302 | 27 | 2 | 524 | 926 | 44 |
| ASV9 | 300 | 30 | 4 | 494 | 936 | 50 |
| ASV10 | 300 | 34 | 2 | 510 | 932 | 44 |
| ASV11 | 14 | 0 | 0 | 511 | 92 | 45 |
| ASV12 | 8 | 0 | 0 | 480 | 79 | 46 |
| ASV13 | 11 | 1 | 0 | 475 | 89 | 42 |
| ASV14 | 8 | 0 | 0 | 491 | 85 | 34 |
| ASV15 | 6 | 3 | 0 | 492 | 101 | 46 |
| ASV16 | 0 | 0 | 0 | 502 | 80 | 52 |
| ASV17 | 1 | 0 | 0 | 485 | 87 | 47 |
| ASV18 | 1 | 0 | 0 | 502 | 97 | 36 |
| ASV19 | 3 | 1 | 0 | 496 | 95 | 44 |
| ASV20 | 2 | 0 | 0 | 473 | 93 | 57 |
OK, everything looks good. This dataset can now be saved in order to use it in other pages as necessary.